The disease and its genetic background
Familial Hypercholesterolaemia (FH) is a rather common inherited disorder of lipid metabolism that affects 1 out of 250 individuals. Each offspring of an affected parent has a 50% risk of inheriting the disease. If both parents are affected, there is a risk of 25% of having a child with the most severe, homozygous form.
FH is caused by mutations in one of these 3 genes: LDLR, APOB or PCSK9 genes. Mutations in these genes affect the clearance of low-density lipoprotein (LDL) by the LDL receptor protein that happens mainly in the liver. LDL is the most abundant and the most atherogenic class of cholesterol-carrying lipoproteins in human plasma, and a reduced clearance leads to elevated blood cholesterol levels in circulation.
FH patients present high blood cholesterol levels since birth, which untreated, represents a very high risk for having a cardiovascular disorder, as a myocardial infarction in early age.
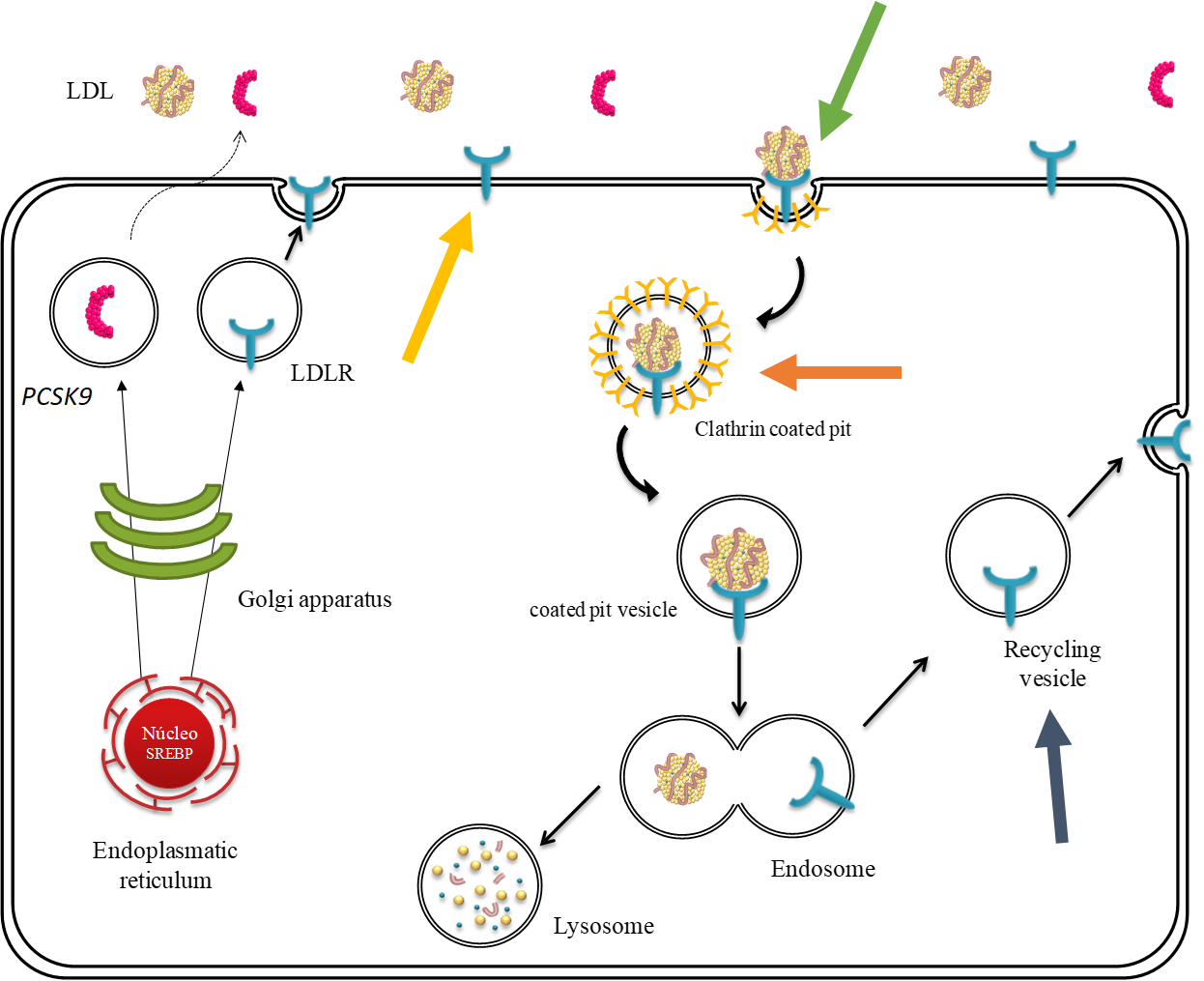
The low densitiy lipoprotein receptor (LDL receptor)) cycle within a liver cell in detail. It shows how the receptor is expressed, transported to the cell membrane where it can bind LDL, then internalised and recycled. All these steps can be affected by variants in the LDLR gene, as indicated by the bold, coloured arrows: LDL receptor expression, binding affinity to LDL, uptake of LDL, recycling of LDL receptor. Pathogenic variants in the APOB gene affect the LDL binding to the LDL receptor and pathogenic variants in the PCSK9 gene affect mostly the recycling of the LDL receptor to the cell surface.
Detailed genetic information is key for a personalised treatment of FH
On average, harbouring a pathogenic variant is associated to a 22 times greater risk of a coronary event than the general population, and 16 times greater than other individuals with identical LDL cholesterol values that do not have FH. However, the exact phenotype is very heterogenous: both the severity of the disease and the response to treatment vary greatly between individual patients. The type of genetic variant found helps predicting the severity of the disorder and can guide the clinician on personalizing the treatment, choosing the best approach for each patient. This is particularly important in children to establish an appropriate, personalised treatment at a young age. If these measures are adopted for lifetime, these patients’ risk for cardiovascular disease will not be elevated at all compared to the general population.

“People with FH profit from early onset of personalised prevention and treatment measures. If diagnosed early by genetic testing and treated properly, these patients will have a controlled clinical situation and will present a cardiovascular risk similar to the general population.”
© private
Mafalda Bourbon, National Institute of Health Doutor Ricardo Jorge; founder and scientific coordinator of the Portuguese FH study.
Therapeutic options for FH patients
There are several therapeutic options to reduce cholesterol levels, some of them more powerful than others: All FH patients need to eat healthy and perform exercise, but for the great majority this is not enough to decrease their elevated cholesterol values. The most commonly used drugs are statins that come with different potency and dosages. In addition, there are two other drugs with different modes of action: Ezetimibe, that has been used in combination with statins for a long time, and a rather new drug, a PCSK9 inhibitor, that can also be given in combination with statins and/or ezetimibe. Further drugs are currently under development.
In detail: How the genotype can help to personalise the treatment regime
The homozygous form of FH, with two affected alleles, is more severe than the heterozygous form, as in the later, one functional copy of the affected gene is still present. In addition, the exact type of gene variant is very important. There are two main groups of variants: One group named “null variants”, the most severe, with no expression of functional LDL receptors from the affected allele at all. The other group is called “defective variants” and these variants allow the expression of an LDL receptor protein that retains some functionality, so these patients are less severely affected and have lower cholesterol values than patients with null allele variants. For a paediatrician, this information is very valuable for determining the age to start treatment and for choosing the best drug or drug combination. Other cardiovascular risk factors can also modulate the phenotype of FH patients, as for example obesity. Inadequate life style habits, as poor diet and smoking, can also have an influence on the patient phenotype and the age of onset of premature cardiovascular disease.
The Portuguese FH study: Results and outlook
The Portuguese FH Study investigates patients for the presence of causative variants in the three genes causing FH and five other genes that are associated with other diseases with very similar clinical presentation. To date, more than 2000 patients have been studied, and a putative pathogenic variant was identified in almost 1000 patients, being about 40% children. From these, 1-2% of all children studied were found not to have FH but another disease of lipid metabolism. Furthermore, the lab also provides the functional characterization of variants that allows us to determine the step in which the LDL receptor cycle is affected by the variant found and how much, if any, residual LDL receptor activity is present.
The Portuguese Familial Hypercholesterolaemia Study was established in 1999 at the National Institute of Health to increase awareness for FH and to implement the molecular diagnosis of FH. A network of 235 clinicians from 84 different hospital departments, public and private, has been established for referral of clinical FH individuals to the Portuguese FH Study during these past 22 years. The study is still ongoing.
So far more than 50 distinct LDLR genetic variants have been characterized by the study. About 80% of all variants found in Portuguese patients have been functionally validated and about 90% were proven to disease causing. This has contributed to the Portuguese FH population having one of the highest rates of definite diagnosis worldwide paving the way for personalized treatment.
Impact of the Portuguese FH study
- Enabling a personalised medicine approach: Early and exact genetic diagnosis is key for tailoring prevention and treatment in FH patients, beginning at a young age.
- Scientific and methodological aspects: The establishment of functional studies and improvement in Next Generation Sequencing (NGS) diagnosis panels, can also contribute to a more personalized medicine, improving patient care and management.
Genetic testing can be done in many labs worldwide and the described treatment options are generally available, so the general principle used here is widely applicable. There are some limitations: Not every lab can perform functional assays, and even if functional assays are done, they are not always sufficient to classify a variant as pathogenic. However, projects are on going to develop a high throughput pipeline to functionally characterise a large number of variants at the same time.
